
Modeling & Simulations
Welcome to the Modeling&Simulations team's webpage. We aim to design high-throughput models for interface-driven properties and processes. Our models cover a wide range of length- and time-scales while being developed and tested with commercial and open-source software on various computing infrastructures from regular workstations to supercomputers. For bridging the scales, we rely on state-of-the-art phenomenological models and theories as well as machine learning techniques, enabling direct comparison of our simulation results with experimental measurements from our collaborators and the literature. To learn more, please check our competencies below.
Ab-initio calculations
Our approach
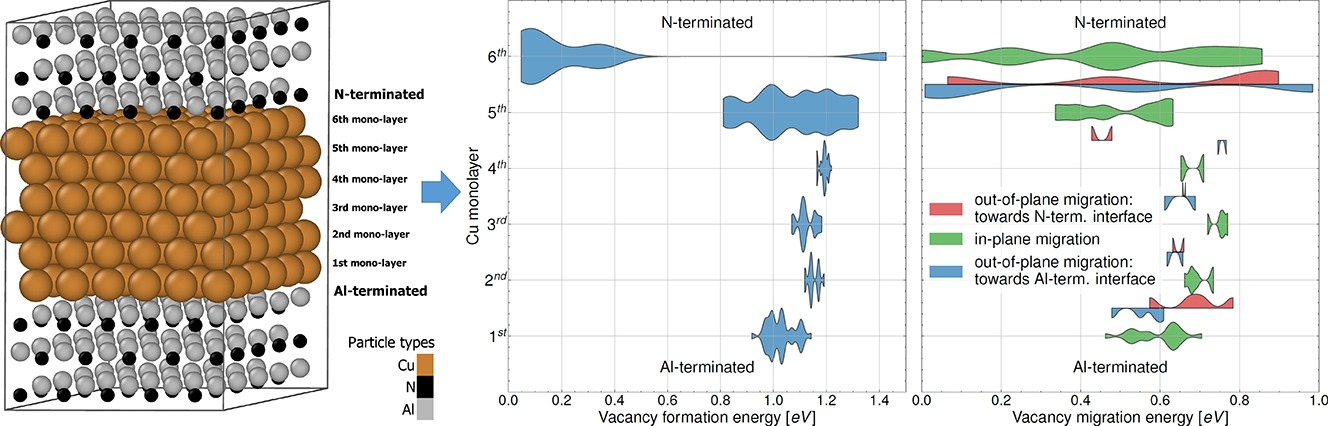
We adopt major open-source and commercial codes for quantum calculations to access various interfacial properties of materials, including interface energy and stress [1], interface vacancy formation energies and migration barriers [2,3], and charge transfer across metallic interfaces to enable high-end interpretation of experimental Auger chemical shifts in X-ray photoelectron spectroscopy [4].
Our tools
- Quantum ESPRESSO - an integrated suite of open-source codes for electronic-structure calculations and materials modeling at the nanoscale based on density-functional theory, plane waves, and pseudopotentials
- CP2K - an open-source quantum chemistry and solid state physics software package providing a general framework for different modeling methods such as DFT using the mixed Gaussian and plane waves approaches
- VASP - a commercial package for performing ab initio quantum mechanical calculations using either Vanderbilt pseudopotentials, or the projector augmented wave method, and a plane wave basis set (licenses available in the group)
Representative publications:
Experimental and ab initio derivation of interface stress in nanomultilayered coatings: Application to immiscible Cu/W system with variable in-plane stress, Applied Surface Science (2024) Explaining the effect of in-plane strain on thermal degradation kinetics of Cu/W nano-multilayers, Scripta Materialia (2024) Anomalously low vacancy formation energies and migration barriers at Cu/AlN interfaces from ab initio calculations, Scripta Materialia (2024) Chemical state analysis of differently miscible interfaces in metallic multilayers, ArXiv (2024)
Upscaling ab-initio with neural network potentials
We adopt major open-source Python tools for the development of new neural network interatomic potentials for upscaling ab-initio calculations, reaching experimentally relevant system sizes as well as enabling chemically accurate large-scale atomistic simulations. For example, we developed new graph neural network potentials for amorphous alumina with various accuracy/performance ratios by recycling an existing ab-initio database, proving their superiority against classical models for non-stoichiometric cases [1]. Using a foundational graph neural network model with Van-der-Waals dispersion correction, we achieved impressive agreement between atomistic simulations, theory, and experiments, uncovering the structure and chemical states of atoms in atomic layer deposited amorphous alumina with varying hydrogen content [2]. On the other hand, we developed chemically-accurate neural network potential for the Cu-W system [3], uncovering the origins of extreme interface stress magnitudes and sign inversion in Cu/W nano-multilayers, with the former attributed to metastable phase formation and the latter to intermixing during magnetron sputtering [4]. We also quantified the effect of in-plane strain, temperature, porosity, grain size, and layer thickness on out-of-plane Young's modulus of Cu/W nano-multilayers, explaining experimental measurements and highlighting the detrimental effect of excess free volume within the Cu and W layers [5].
Our tools
- n2p2 - Behler-Parinello neural network potential development package
- NequIP- data-efficient graph neural network potential development package
- Matlantis - foundational Preferred Networks (PFN)’s proprietary neural network potential for 72 elements
Representative publications:
Do we really need machine learning interatomic potentials for modeling amorphous metal oxides? Case study on amorphous alumina by recycling an existing ab initio database, Modelling and Simulation in Materials Science and Engineering (2024) Interpreting Auger parameter shifts for atomic layer deposited amorphous alumina with varying hydrogen content: Insights from atomistic simulations, ab initio calculations and electrostatic theory of extra-atomic relaxation, ArXiv (2024) Machine learning interatomic potential for Cu/W composites, ArXiv (2024) Uncovering the origin of interface stress enhancement and sign inversion in immiscible nanomultilayers, ArXiv (2024) Effect of internal stress and microstructure on mechanical properties of sputter-grown Cu/W nanomultilayers, ArXiv (2024)
Large-scale atomistic simulations
We adopt major open-source community codes for large-scale atomistic simulations with millions of atoms to model nanoscale phenomena. More specifically, we operate with the following methods:
- The classical molecular dynamics (MD) method provides us with access to ultra-fast interface-driven processes at the nanoscale. Successful examples include self-propagating high-temperature synthesis (SHS) in reactive Ni/Al nano-multilayers [1,2], fast interfacial diffusion, premelting, and melting point depression in non-reactive Cu/AlN nano-multilayers [3], temperature-induced amorphization, surface segregation, and core stress state transition in alumina nanoparticles [4], as well as rapid solidification and heterogeneous nucleation in Cu matrix composites reinforced with W nanoparticles [5].
- The hybrid molecular dynamics/Monte Carlo (MD/MC) method provides us with access to nanoscale thermodynamics at elevated temperatures, for example, uncovering defect-phases (so-called complexions) in metal alloys. These include planar grain boundary complexions and amorphous intergranular films [6], as well as linear complexions at dislocations [7,8].
- The non-equilibrium molecular dynamics (NEMD) method provides us with access to nanoscale mechanics, including dislocation-driven plasticity in nanostructured materials [6,9,10].
Currently, our efforts are focused on developing new competencies in diffusive molecular dynamics and kinetic Monte Carlo techniques, to expand the timescales of our large-scale atomistic simulations beyond the capabilities of traditional methods listed above, enabling direct comparison with in-situ experimental characterization of diffusion-driven processes.
Our tools
- LAMMPS - a high-performance atomistic modeling code enabling large-scale atomistic simulations with millions or even billions of atoms
- OVITO - a visualization and analysis software for output data generated in atomistic simulations
Representative publications:
Alloying propagation in nanometric Ni/Al multilayers: A molecular dynamics study, Journal of Applied Physics (2017) Microstructure evolution and self-propagating reactions in Ni-Al nanofoils: An atomic-scale description, Journal of Alloys and Compounds (2017) Atomistic assessment of melting point depression and enhanced interfacial diffusion of Cu in confinement with AlN,ACS Applied Materials & Interfaces (2022) Atomistic simulations of the crystalline-to-amorphous transformation of γ-Al2O3 Nanoparticles: Delicate Interplay between lattice distortions, stresses, and space charge, Langmuir (2023) Directional solidification of Cu with dispersed W nanoparticles: A molecular dynamics study in the context of additive manufacturing, Materialia (2024) Grain boundary complexions and the strength of nanocrystalline metals: Dislocation emission and propagation, Acta Materialia (2018) Linear complexions: Metastable phase formation and coexistence at dislocations, Physical Review Letters (2019) Prediction of a wide variety of linear complexions in face centered cubic alloys, Acta Materialia (2020) Interdependent linear complexion structure and dislocation mechanics in Fe-Ni, Crystals (2020) Linear complexions directly modify dislocation motion in face-centered cubic alloys, Materials Science and Engineering: A (2020)

Simulation-guided phenomenological modeling and machine learning
We rely on well-established theories and phenomenological modeling to analyze and interpret our atomistic modeling results as well as guide the development of new theories and mesoscale modeling tools, enabling direct comparison with experiment. For example, we pioneered the development of diffusion-driven dissolution [1] and dissolution-driven reactive wave propagation [2] models for reactive nano-multilayers, described disconnection-driven growth of twin embryos in hexagonal close-packed metals [3], and more recently, refined electrostatic model of extra-atomic relaxation for simple interpretation of Auger parameter shifts in X-ray photoelectron spectroscopy of crystalline and amorphous materials [4].
The latter is an essential part of our ongoing efforts for bridging high-quality atomistic simulations and experimental characterization, through extensive use of theory as well as state-of-the-art machine learning tools to address the most pressing questions in interface science. In particular, Machine Learning has been found particularly useful in extracting macroscopic features from noisy atomistic simulations, relating, for example, stress signatures with particular twin/matrix interfaces in Mg-Al alloys, enabling direct comparison with experiment [5].
Representative publications:
- Dissolution process at solid/liquid interface in nanometric metallic multilayers: Molecular dynamics simulations versus diffusion modeling, Acta Materialia (2015)
- Modeling self-sustaining waves of exothermic dissolution in nanometric Ni-Al multilayers, Acta Materialia (2016)
- Disconnection-mediated twin embryo growth in Mg, Acta Materialia (2020)
- Interpreting Auger parameter shifts for atomic layer deposited amorphous alumina with varying hydrogen content: Insights from atomistic simulations, ab initio calculations and electrostatic theory of extra-atomic relaxation, ArXiv (2024)
- Machine learning of twin/matrix interfaces from local stress field, Computational Materials Science (2023)
Multi-physics models (for laser processing)
Our approach
We adopt the Eulerian-Lagrangian framework to numerically describe the multiphysics nature of laser processing of metals. We use
-
Computational Fluid Dynamics (CFD) based on Finite Volume Method (FVM) to model heat and mass transport in each phase as well as solid-liquid phase transitions in the substrate
-
Volume-of-Fluid (VOF) method to model solid-gas and liquid-gas interfaces, involving multiphase flow
-
Discrete Element Method (DEM) to model powder particles and laser photons and their interactions
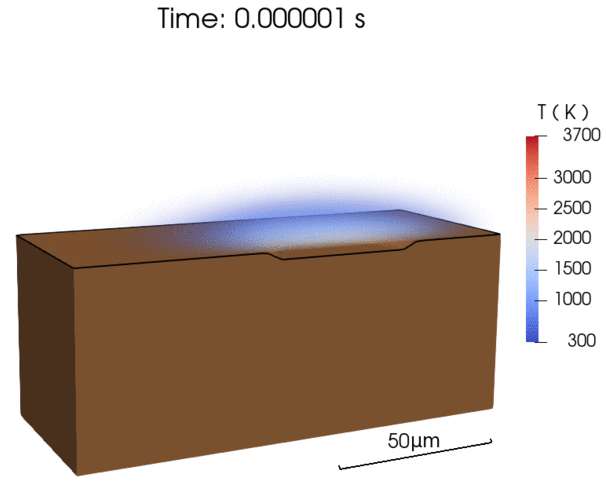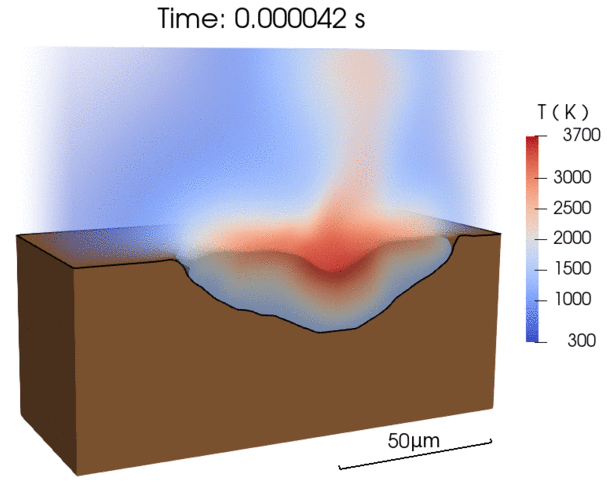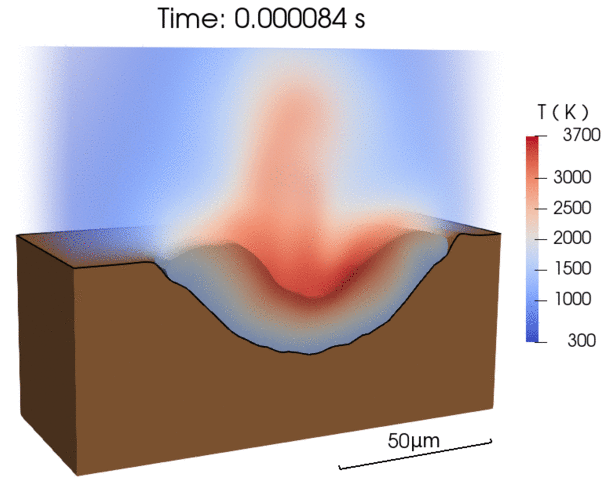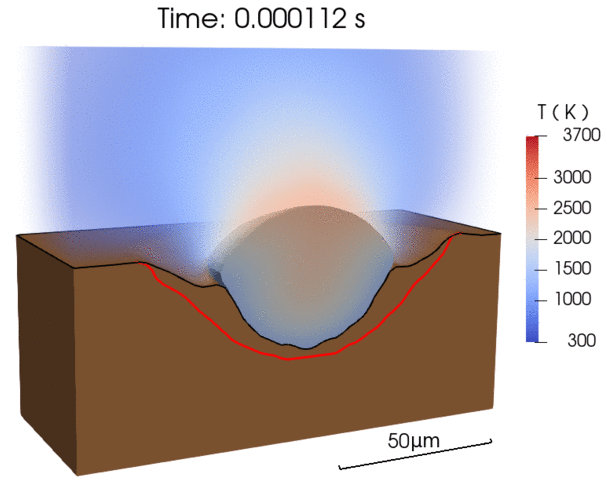
Recent efforts were focused on enabling energy-efficient laser processing of copper by (1) predicting light absorption on rough surfaces and sparse powder beds [1], (2) uncovering in-situ nanoparticle deposition from the vapor plume during microwelding [2], as well as (3) explaining balling defect formation mechanisms during laser powder bed fusion [3]. By combining our modeling results with experimental characterization, we derive simplified models that can be applied by the research community to a wide range of materials [1,3].
Our tools
-
OpenFOAM - open-source FVM-based software designed for CFD simulations
-
Aspherix - commercial DEM software designed for modeling granular media
-
CFDEM coupling - commercial software designed for modeling particle-fluid interactions with partially open code allowing for customized subroutines for CFD-DEM coupling using OpenFOAM and Aspherix
-
ParaView - open-source software used for post-processing and visualization of simulation results from software listed above
Representative publications
- Modeling of laser beam absorption on rough surfaces, powder beds and sparse powder layers, SSRN (2024)
- Energy-efficient microwelding of copper by continuous-wave green laser: insights into nanoparticle-assisted absorptivity enhancement, SSRN (2024)
- A simple scaling model for balling defect formation during laser powder bed fusion, Additive Manufacturing (2023)
Current and former team members
From left to right: Vladyslav Turlo, Javier Fernandez Troncoso, Yann Muller, Giandomenico Lupo, Simon Gramatte, Stephane Nilsson, Stefan Scharen
August 2022 - Thun
Team leader

Curriculum Vitae
Scientists

Dr. Manura Liyanage
Expert in machine learning interatomic potential (MLIP) development
Project: Neural network potential development for Cu/W nanocomposites

Dr. Yang Hu
Expert in large-scale atomistic simulations
Project: Atomistic modeling of heterogeneous interfaces
Ph.D. students

Project: Atomistic modeling of systems with complex chemical bonding
Finished projects and alumni
Postdoctoral projects
- Dr. Giandomenico Lupo (2020-2022): Multiphysics modeling of laser-matter interactions
- Dr. Javier Fernandez Troncoso (2021-2022): Ab initio modeling of Cu/W interfaces
- Dr. Javier Fernandez Troncoso (2020-2021): Atomistic modeling and machine learning for Mg research
PhD-related projects
- Simon Gramatte (2020-2021): Atomistic study of solid/liquid interface mobility
Master theses
- Yann Muller (2021): Molecular dynamics simulations of Cu/AlN nano-multilayers
Internships
- Stefan Scharen (2022): Atomistic simulations of heterogeneous nucleation
- Yann Muller (2022): Ab initio modeling of Cu/AlN nano-multilayers
- Stephane Nilsson (2021): Bayesian inference for multiphysics models of laser melting
- Daniele Hamm (2021): Bayesian inference for heat transfer models of laser melting
- Jose Simon Greminger (2021): Atomistic modeling of heterogeneous nucleation
- Jose Simon Greminger (2020): Atomistic modeling of alumina nanoparticles